Фенилкетонурия – основные симптомы:
- Кожная сыпь
- Рвота
- Судороги
- Потливость
- Сонливость
- Раздражительность
- Запор
- Задержка в развитии
- Умственная отсталость
- Плаксивость
- Расстройство мышечного тонуса
- Дрожание
- Вялость
- Нарушение равновесия
- Синюшность конечностей
- Дерматит
- Уменьшение размеров головы
Что такое фенилкетонурия
Фенилкетонурия представляет собой достаточно тяжелое наследственное заболевание, основная тяжесть проявлений которого сосредоточена, прежде всего, на нервной системе. Фенилкетонурия, симптомы которой чаще всего встречаются среди девочек, возникает по причине нарушения обмена аминокислот, учитывая же поражение при этом ЦНС, ее проявления сводятся к нарушению умственного развития.
- Описание заболевания
- Cимптомы фенилкетонурии
- Диагностирование фенилкетонурии
- Причины фенилкетонурии
- Лечение фенилкетонурии
Общее описание
В классической своей форме, которая актуальна для большинства случаев, фенилкетонурия, которую также принято определять как фенилпировиноградную олигофрению, связана с резкостью снижения активности печеночного фермента. Им в частности является фениланин-4-гидроксилаз. Нормальное состояние организма предусматривает катализацию им превращения в тирозин фенилаланина.
1% случаев отмечается возникновением атипичной формы фенилкетонурии, которая возникает по причине мутаций в другого типа генах, которые ответственны за процесс кодирования ферментов. Наследование заболевания происходит в соответствии с аутосомно-рецессивной схемой.
Что касается процессов, происходящих при рассматриваемом заболевании, то они заключаются в следующем. Возникновение характерного метаболического блока провоцирует активацию побочных путей по обмену фенилаланина, что приводит к накоплению токсичных производных от его действия. К таким производным в частности относятся фенилмолочная и фенилпировиноградные кислоты, практически не образующиеся при нормальном состоянии организма.

Они же влияют на ЦНС, провоцируя нарушения белкового обмена, обмена липопротеидов и обмена гликопротеидов. Одновременно с этим возникают расстройства в транспорте аминокислот, нарушения в обмене серотонина и катехоламинов, актуальность приобретают также и перинатальные факторы.
Помимо этого начинают также образовываться и в норме практически отсутствующие ортофенилацетат и фенилэтиламин. Их избыток провоцирует нарушения в метаболизме липидов, происходящем в головном мозге. Как предполагается, именно это становится причиной прогрессирующего состояния снижения у больных данным заболеванием интеллекта, что может достичь даже идиотии.
В целом же окончательно механизм, в соответствии с действием которого происходит нарушение в развитии функций мозга в случае с актуальным для организма заболеванием фенилкетонурией, не ясен.
Фенилкетонурия: симптомы заболевания
Здесь, прежде всего, важно отметить, что первые недели жизни ребенка не позволяют внешне определить данного заболевания. Первые его признаки проявляются через два-шесть месяцев с момента рождения малыша. Он становится вялым, наблюдается отсутствие заинтересованности в отношении условий, его окружающих и мира в целом. Также ребенок становится беспокойным, нарушениям подвергается мышечный тонус.
Появляется рвота, судороги, тяжелые кожные экземы. Под экземами в частности подразумевается острая или хроническая форма воспалительного и незаразного заболевания, при котором образуется сыпь. Природа заболевания аллергическая, дополнительными проявлениями симптоматики выступает чувство жжения и выраженный зуд кожи. Актуальна и склонность к рецидивам, то есть к повторному возникновению симптоматики после относительного временного его затишья.
Шестой месяц позволяет определить отставание в развитии у ребенка. Одновременно с этим теряется способность к фокусированию взгляда на отдельных предметах, малыш перестает узнавать родителей. Отсутствует реакция на цветные/яркие игрушки. Важно оперативно приступить к лечению, в противном случае отсталость развития будет постепенно подвергаться лишь прогрессированию в актуальных для нее процессах.

Физическое развитие больных младенцев на физическом уровне отмечается меньшими нарушениями, нежели на психическом. В обхвате голова может быть несколько меньших размеров, чем это предусмотрено для показателей нормы. Зубки прорезываются позже, позже ребенок начинает сидеть и ходить.
Принятие положения стоя у таких малышей сопряжено с широким расставлением для этого ног, а также со сгибанием их в коленях и в тазобедренных суставах, плечи и голова при этом опущены. Что касается особенностей ходьбы у больных детей, то она характеризуется покачиваниями, шажки небольшие. Сидят дети, поджав под себя ножки, что обуславливается значительным мышечным тонусом, который они испытывают.
Отличаются детки и характерной внешностью со светлыми волосами, кожа у них абсолютно белая, без пигментации, глаза светлые. Учитывая излишнюю белизну кожи, она нередко покрывается у детей сыпью, что объясняется ее особой чувствительностью в отношении воздействия ультрафиолетового излучения.
В числе основных проявлений, свойственных фенилкетонурии, можно также выделить характерный «мышиный» запах, в некоторых случаях возможны эпилептические припадки, которые, однако, с возрастом исчезают. Выраженными проявлениями выступают синюшность конечностей, дермографизм (изменение в окраске кожи местного типа, происходящее при механическом воздействии на нее), потливость.
Чаще среди больных фенилкетонурией отмечается, помимо перечисленных симптомов, наличие артериальной гипотонии, дерматита, частых запоров, тремор (т.е. дрожание), потеря равновесия и расстройство в виде нарушения координации движений.
Диагностирование фенилкетонурии
Важным, как мы уже отметили, является раннее диагностирование заболевания, что позволит избежать его развития и привести к ряду необратимых и тяжелых последствий. По этой причине в родильных домах к 4-5 дням жизни (для новорожденных доношенных) берется для анализа кровь. У недоношенных детей на предмет фенилкетонурии (ФКУ) кровь берется на 7 день.
Процедура предусматривает взятие капиллярной крови по прошествии часа с момента кормления, ею в частности пропитывается специальный бланк. Концентрация, указывающая на отметку свыше 2,2% фенилаланина в крови малыша, требует направления его с родителями для осмотра в медико-генетический центр. Там же проводится дообследование и, собственно, уточнение диагноза.
Причины фенилкетонурии
Фенилкетонурия может быть спровоцирована следующими факторами:
- Близкородственные браки, при которых, помимо иных патологий, повышается вероятность рождения ребенка с этим заболеванием;
- Мутация гена (т.е. его изменение), произошедшая по тем или иным причинам в области локализации 12 хромосомы.
Сам процесс наследования гена ФКУ может носить случайный характер, что в качестве примера отражено в приведенной ниже схеме.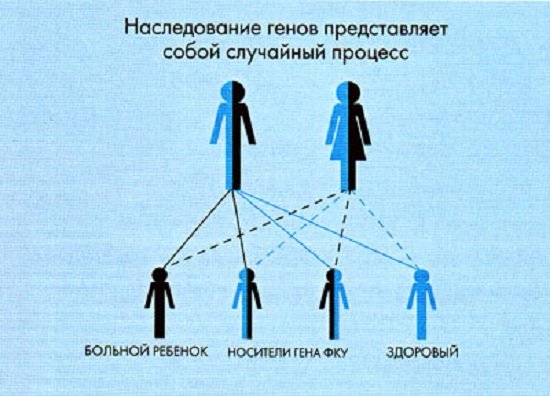 Возможность наследования генов ФКУ
Возможность наследования генов ФКУ
Лечение фенилкетонурии
Единственный метод лечения заключается в своевременной организации диетотерапии, которая требуется с первых дней жизни. Заключается она в резком ограничении фенилаланина, который содержится в отдельных пищевых продуктах. Таким образом, исключаются все белковые продукты.
Учитывая продолжительность и полное исключение фенилаланина из пищи, возможным становится расщепление собственных белков, что ведет к истощению организма больного. По этой причине потребность в получении белка возмещается за счет аминокислотных смесей и белковых гидролизатов.
Со сменой снижения концентрации в крови фенилаланина до нормальных показателей, в рацион постепенно добавляются продукты животного происхождения. В рацион добавляются различные фрукты и сезонные овощи, растительные и животные жиры, а также углеводы. Естественно, что контроль над содержанием фенилаланина следует продолжать вести. Организм должен быть снабжен данной аминокислотой в должном объеме для обычного развития и роста малыша, однако, за исключением возможности скапливания его в тканях.
Строжайшая диета соблюдается на протяжении, как минимум, пяти лет. Что касается возраста более взрослого, то здесь в значительной степени снижается подверженность нервной системы к воздействию, оказываемому фенилаланином, а также воздействию, оказываемому продуктами его распада. При соблюдении требуемых мер к 12-14 годам ребенок сможет свободно перейти к обычному питанию.
Примечательно, что медикаментозное лечение при данном заболевании носит синдромный характер. В него включено применение препаратов, ориентированных на устранение судорог, в том числе и препаратов, которые оказывают стимулирующее воздействие на интеллектуальную деятельность. В обязательном порядке детям назначается курс лечебной физкультуры и массажа. Дополнительно назначаются уроки, способствующие развитию логики.
Диагностирование фенилкетонурии производится педиатром и генетиком на основании специальных анализов в комплексе с общей характерной симптоматикой.
Что делать?
Если Вы считаете, что у вас Фенилкетонурия и характерные для этого заболевания симптомы, то вам могут помочь врачи: педиатр, генетик.
Фенилкетонурия у детей
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты.
Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез фенилкетонурии
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов — фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко — врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика фенилкетонурии
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным.
Скрининг-тест проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.

Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др.
Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Прогноз и профилактика фенилкетонурии
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием.
Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода.
Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Понравилась статья? Поделись с друзьями в соц.сетях:


